
以Jens K. Nørskov为代表提出计算氢电极模型的固定电子数电化学模拟方法通过电化学反应中间体在电中性催化剂上的吸附自由能,再加上电能修正项(-eU)和pH值修正项来研究催化剂活性。该模型通过简单的催化模拟流程成功预言了一系列性能优异的催化剂,因此被电化学研究者广泛采用。以该模型为基础,经过20多年达发展,电化学模拟逐渐形成通过描述符高通量筛选催化剂,与机器学习相结合设计催化剂的电催化理论计算框架。但该模型忽略了电化学环境下电势控制电极材料的实际带电状态及该带电状态对反应活性的影响。针对上述问题,近年来包括Jens K. Nørskov课题组、William A. Goddard III课题组、Martin P. Head-Gordon课题组和Ravishankar Sundararaman课题组等著名的电化学计算模拟组采用“先结构优化后电子数调整分步走”的策略来修正上述影响(电子数优化流程见图2中蓝色流程部分)。然而,由于电荷变化引起体系能量的剧烈波动~∆qUabs(Uabs为绝对电势),该方法存在能量难以收敛的问题,导致计算成本昂贵。与此同时,由于模拟过程中无法实现结构和电子数的同步调整,严重制约了其应用范围,只能实现静态自洽计算功能。
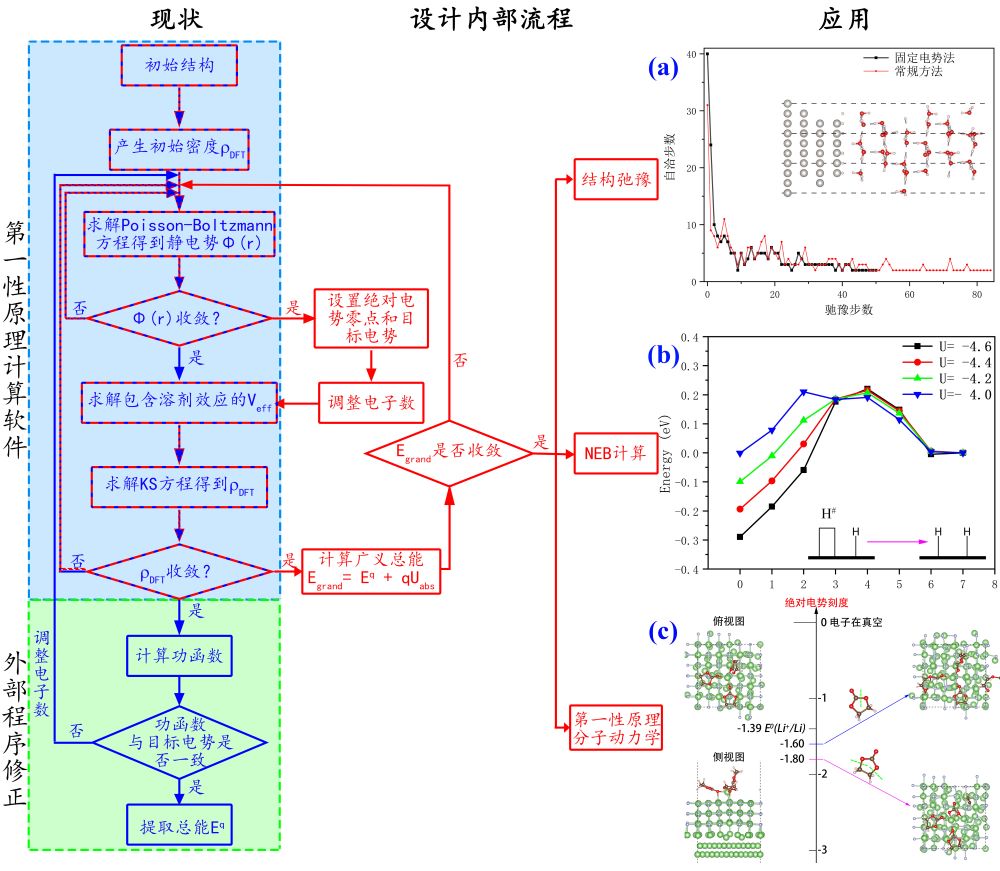
申请人和博后导师汪林望教授针对上述瓶颈,在第一性原理软件PWmat的基础上开发了巨正则固定电势法。该方法通过修改第一性原理计算内部流程(见图中红色流程部分),将设定参考电势和目标电势、调整体系电子数和以广义能量作为收敛对象这些功能直接融入第一性原理计算的SCF自洽计算过程中,采用“结构优化和电子数优化的同步走”策略,实现了恒电势下第一性原理的结构优化、过渡态搜索、分子动力学等功能,突破了固定电子数法电化学模拟技术瓶颈,建立了以绝对电势为参考系的恒电势电化学计算模拟技术方法体系。且所需计算成本和常规方法一致(见图a)。
虽然目前的固定电势法只能实现电子的巨正则,还无法实现离子的巨正则,该方法已经集成到商业化的第一性原理软件PWmat中,是电化学模拟领域世界领先的技术。使用和发展固定电势法有望重塑现有的电催化理论计算框架。
基于该方法发表的文章:
1. G. Gao*, L. Wang*. Substantial potential effects on single-atom catalysts for the oxygen evolution reaction via the fixed-potential method. J. Catal. 2020, 391, 530-538.
2. G. Gao*, L. Wang*. A potential and pH inclusive microkinetic model for hydrogen reactions on Pt surface.Chem Catal. 2021, 1, 1331-1345.
3. Q. Zhang, Y. Zhang, G. Gao*, S. Zhang*. Potential-driven semiconductor-to-metal transition in monolayer transition metal dichalcogenides. Adv. Funct. Mater. 2023, 33, 2208736.
4. Q. Zhang, Y. Zhang, S. Zhang*, G. Gao*. Potential effects on the catalytic mechanisms of OER and ORR. J. Phys. Chem. C 2023,127, 16346-16356.
5. G. Gao*, L. Wang*. The concerted proton-electron transfer mechanism of proton migration in the electrochemical interface. iScience. 2023, 26, 108318.
6. G. Gao*, L. Wang*. Protocol for evaluating the effect of the potential on electrochemical reaction via the grand canonical fixed-potential technique. STAR Protoc. 2024, 5, 103021.
 (创新港)
(创新港)


