|
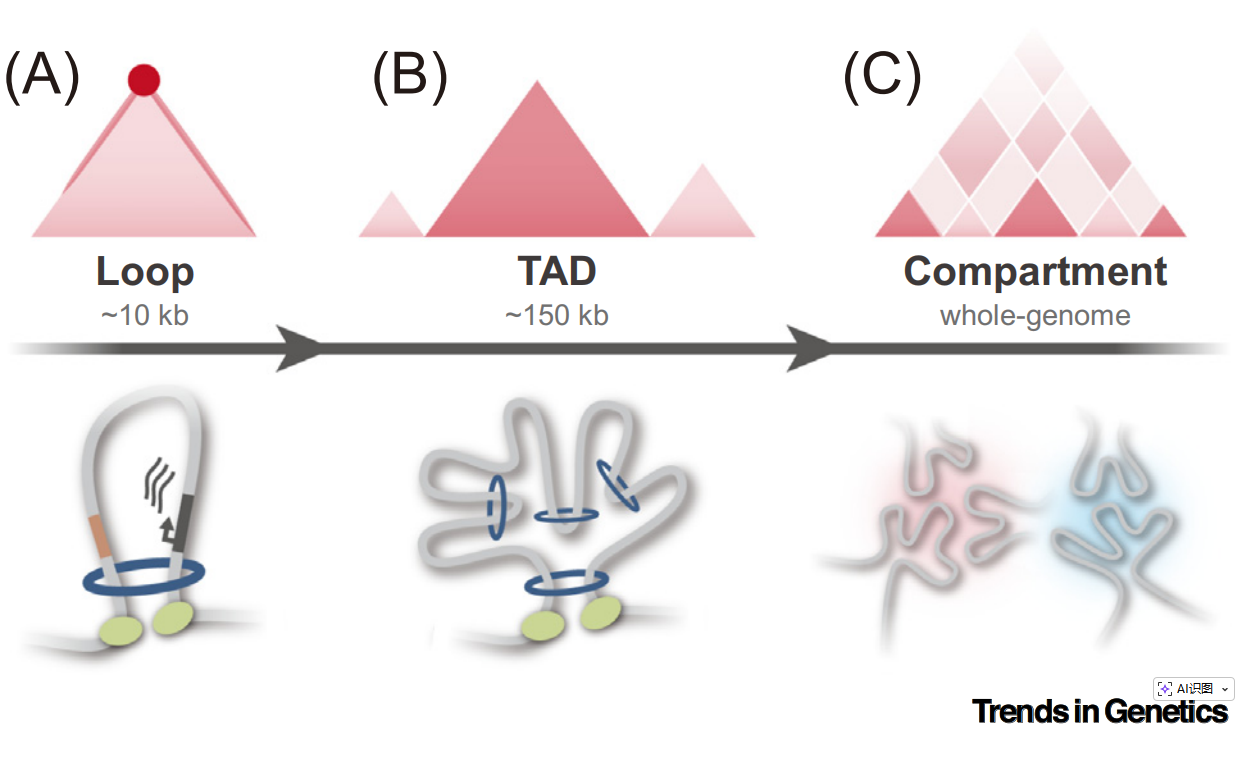
Multiscale evolution of the 3D genome Yizhuo Che , Stephen J. Bush , Kai Ye Trends in Genetics . 345–356 (2026) . [PDF] The spatial organization of the genome underpins how and when its encoded information is utilized. Nevertheless, a unified understanding of how genome architecture evolves across different life scales is still emerging. This review synthesizes current research on 3D genome evolution from both micro- and macro-perspectives. At cellular-level timescales exemplified by cancer, architectures can evolve rapidly due to frequent genomic mutations and plastic epigenetic marks. Between closely related species, architectural divergence is driven primarily by local genomic alterations, mainly cis-regulatory elements. Across larger phylogenetic distances, genome architectures display striking diversity, yet recurrent higher-order features have nevertheless emerged through convergent evolution, reflecting common functional requirements. The micro-to-macro comparative framework proposed here delineates how the diversity of genome architectures relates to the evolution of form and function. |
 |
The evolution of high-order genome architecture revealed from 1,000 species Yizhuo Che, Stephen J. Bush, Hui Lin, Mingxuan Li, Xiaofei Yang, Qi Xie, Yuchun Liu, Deyu Meng, Kai Ye Cell . (2026) . [PDF] Spatial genome organization plays a crucial regulatory role, but its evolutionary development remains unclear. Leveraging Hi-C data from 1,025 species, we trace the evolutionary trajectories of genome organization through 2 higher-order architectures, “global folding” (spatial organization of the karyotype) and “checkerboard” (spatial organization of chromatin compartments). Earlier unicellular life forms mostly displayed random genome configurations. Throughout the evolution of plants, global folding became and remained the prominent architecture. However, animals progressively developed more pronounced checkerboard architectures; these are also apparent during early embryogenesis, which suggests that they act as a conserved mechanism of gene regulation. In contrast, plants exhibit comparatively weaker checkerboard patterns and instead preferentially organize co-regulated genes into linear genomic clusters. Both strategies of gene arrangement reinforce the biological principle that “structure determines function”: divergent evolutionary paths converge on ...... |
 |
SpatialCOC: an integrative framework for spatial continuous mapping and cross-omics correction in spatial multi-omics data Mingxuan Li, Peisen Sun, Yisi Luo, Guancheng Zhou, Xiaofei Yang, Deyu Meng, Kai Ye Nature Communications . (2026) . [PDF] Integrating spatial multi-omics data presents significant challenges, particularly in uncovering the spatial patterns of cells and deciphering the real regulatory mechanisms among various omics. These insights are critical for harnessing the full potential of each modality while minimizing the impact of biotechnological biases that will lead to unstable results. Here, we introduce SpatialCOC, a framework that treats spatial information as prior knowledge to learn omics-specific spatial distributions, then discovering nonlinear correlations among modalities. The effectiveness and robustness of SpatialCOC are validated using real-world datasets, encompassing diverse tissue sections analyzed with multiple experimental techniques. Compared to existing methods, SpatialCOC excels in identifying region-specific continuous spatial domains and maintains batch-consistency across trajectory inferences. By providing a novel perspective on the interplay between spatial information and multi-omics modalities, SpatialCOC offers a flexible approach that can accommodate modality data of arbitrary dimensions. |
 |
Highly accurate ab initio gene annotation with ANNEVO Pengyu Zhang, Tun Xu, Songbo Wang, Xiaofei Yang, Peisen Sun, Peng Jia, Jiadong Lin, Bo Wang, Yizhe Zhang, Deyu Meng, Stephen J. Bush, Zemin Ning, Kai Ye Nature Methods. 23, 740–748 (2026). [PDF] Accurate gene annotation is essential for deciphering the mapping from genomic sequences to their functional roles. However, current methods struggle to model complex gene transmission patterns, such as vertical inheritance and horizontal gene transfer. Here we introduce ANNEVO, a mixture of experts-based genomic language model that directly models distal sequence dependencies and joint evolutionary relationships from diverse genomes, enabling precise ab initio gene annotation. Through extensive benchmarking on 566 phylogenetically diverse species, we demonstrate that ANNEVO substantially outperforms existing ab initio methods and achieves performance comparable to state-of-the-art annotation pipelines. Furthermore, ANNEVO’s independence from external evidence allows it to deliver more complete annotations than reference annotations for a broad range of ...... |
 |
Population-level structural variant characterization using pangenome graphs Songbo Wang, Tun Xu, Pengyu Zhang, Kai Ye Nature Genetics. 58, 664–672 (2026). [PDF] Population-level structural variant (SV) profiling is crucial in the era of pangenomes. However, identifying SVs from genome assemblies and pangenome graphs remains a substantial challenge. Here we present Swave, a sequence-to-image, deep learning-based method that accurately resolves both simple and complex SVs, along with their population characteristics, from assembly-derived pangenome graphs. Swave introduces ‘projection waves’ to summarize the dotplot images that capture mapping patterns between reference and SV-indicating alleles in the pangenome. Then, a recurrent neural network distinguishes true SV signals from background noise introduced by genomic repeats. Swave demonstrates superior performance in both SV-type classification and genotyping compared with existing methods. When applied to healthy cohorts and rare-disease cohorts, Swave reveals ......
|
 |
Spatially resolved single-cell transcriptome analysis of murine Salmonella infection reveals the role of distal colonocytes in the inflammatory response Dan Xu ,Ruifen Zhang ,Shanshan Li ,Can Guo ,Chenglin Guan ,Xiang Li ,Mengyao Guo ,Xin Xu ,Yaxin Liu ,Chenyi Mao ,Peisen Sun ,Xiaomin Dang ,Diya Sun ,Chengyao Wang ,Stephen J. Bush ,Kai Ye Gut Microbes. Dec 31;17(1):2579909 (2025) . [PDF] The intestine is a highly compartmentalized organ, with distinct segments exhibiting both varying susceptibilities and responses to enteric pathogens, although the cellular and molecular bases of these responses remain elusive. Here, we used Salmonella Typhimurium (S. Tm), a prominent enteric pathogen that causes human colitis, to establish a murine model of Salmonella enterocolitis. By integrating bulk RNA-seq, single-cell RNA-seq, and spatial RNA-seq data, we present a comprehensive spatiotemporal single-cell transcriptomic landscape of the colon over a week-long time course of infection. We identified the distal colon as the intestinal segment where most of the host responses were initiated, with distal colonocytes (DCCs) being the most responsive epithelial cells upon the onset of infection. Furthermore, by correlating our findings with human intestinal single-cell transcriptome data, we identified a human colonocyte population that shares many characteristics with murine DCCs. Our study advances the understanding of the cellular and molecular basis of compartmentalized intestinal responses to pathogenic insults and may pave the way for novel preventive and therapeutic strategies to mitigate intestinal damage and combat intestinal infections. |
 |
Deciphering Complex Interactions Between LTR Retrotransposons and Three Papaver Species Using LTR_Stream Tun Xu, Stephen J Bush, Yizhuo Che, Huanhuan Zhao, Tingjie Wang, Peng Jia, Songbo Wang, Peisen Sun, Pengyu Zhang, Shenghan Gao, Yu Xu, Chengyao Wang, Ningxin Dang, Yong E Zhang, Xiaofei Yang, Kai Ye Genomics Proteomics Bioinformatics. Sep 22;23(4):qzaf061 (2025) . [PDF] Long terminal repeat retrotransposons (LTR-RTs), a major type of class I transposable elements, are the most abundant repeat element in plants. The study of the interactions between LTR-RTs and the host genome relies on high-resolution characterization of LTR-RTs. However, for non-model species, this remains a challenge. To address this, we developed LTR_Stream for sublineage clustering of LTR-RTs in specific or closely related species, providing higher precision than current database-based lineage-level clustering. Using LTR_Stream, we analyzed Retand LTR-RTs in three Papaver species. Our findings show that high-resolution clustering reveals complex interactions between LTR-RTs and the host genome. For instance, we found that autonomous Retand elements could spread among the ancestors of different subgenomes, like retroviral pandemics, enriching genetic diversity. |
 |
STMiner: gene-centric spatial transcriptomics for deciphering tumor tissues Peisen Sun, Stephen J Bush, Songbo Wang, Peng Jia, Mingxuan Li, Tun Xu, Pengyu Zhang, Xiaofei Yang, Chengyao Wang, Linfeng Xu, Tingjie Wang, Kai Ye Cell Genomics. Feb 12;5(2):100771 (2025) . [PDF] Analyzing spatial transcriptomics data from tumor tissues poses several challenges beyond those of healthy samples, including unclear boundaries between different regions, uneven cell densities, and relatively higher cellular heterogeneity. Collectively, these bias the background against which spatially variable genes are identified, which can result in misidentification of spatial structures and hinder potential insight into complex pathologies. To overcome this problem, STMiner leverages 2D Gaussian mixture models and optimal transport theory to directly characterize the spatial distribution of genes rather than the capture locations of the cells expressing them (spots). By effectively mitigating the impacts of both background bias and data sparsity, STMiner reveals key gene sets and spatial structures overlooked by spot-based analytic tools, facilitating novel biological discoveries. The core concept of directly analyzing overall gene expression patterns also allows for a broader application beyond spatial transcriptomics, positioning STMiner for continuous expansion as spatial omics technologies evolve. |
 |
Shigella infection is facilitated by interaction of human enteric α-defensin 5 with colonic epithelial receptor P2Y11 Dan Xu, Mengyao Guo, Xin Xu, Gan Luo, Yaxin Liu, Stephen J. Bush, Chengyao Wang, Tun Xu,..., Yaming Jiu, Nathalie Sauvonnet, Wuyuan Lu, Philippe J. Sansonetti, Kai Ye Nature Microbiology. 10, 509–526 (2025) . [PDF] Human enteric α-defensin 5 (HD5) is an immune system peptide that acts as an important antimicrobial factor but is also known to promote pathogen infections by enhancing adhesion of the pathogens. The mechanistic basis of these conflicting functions is unknown. Here we show that HD5 induces abundant filopodial extensions in epithelial cells that capture Shigella, a major human enteroinvasive pathogen that is able to exploit these filopodia for invasion, revealing a mechanism for HD5-augmented bacterial invasion. Using multi-omics screening and in vitro, organoid, dynamic gut-on-chip and in vivo models, we identify the HD5 receptor as P2Y11, a purinergic receptor distributed apically on the luminal surface of the human colonic epithelium. Inhibitor screening identified cAMP-PKA signalling as the main pathway mediating the cytoskeleton-regulating activity of HD5. In illuminating this mechanism of Shigella invasion, our findings raise the possibility of alternative intervention strategies against HD5-augmented infections. |
 |
The centromere landscapes of four karyotypically diverse Papaver species provide insights into chromosome evolution and speciation Shenghan Gao , Yanyan Jia , Hongtao Guo , Tun Xu , Bo Wang , Stephen J. Bush , Shijie Wan , Yimeng Zhang , Xiaofei Yang , Kai Ye National Science Review. 4(8):100626 (2024) . [PDF] Understanding the roles played by centromeres in chromosome evolution and speciation is complicated by the fact that centromeres comprise large arrays of tandemly repeated satellite DNA, which hinders high-quality assembly. Here, we used long-read sequencing to generate nearly complete genome assemblies for four karyotypically diverse Papaver species, P. setigerum (2n = 44), P. somniferum (2n = 22), P. rhoeas (2n = 14), and P. bracteatum (2n = 14), collectively representing 45 gapless centromeres. We identified four centromere satellite (cenSat) families and experimentally validated two representatives. For the two allopolyploid genomes (P. somniferum and P. setigerum), we characterized the subgenomic distribution of each satellite and identified a “homogenizing” phase of centromere evolution in the aftermath of hybridization. An interspecies comparison of the peri-centromeric regions further revealed extensive centromere-mediated chromosome rearrangements...... |
 |
Near telomere-to-telomere genome assemblies of two Chlorella species unveil the composition and evolution of centromeres in green algae Bo Wang, Yanyan Jia, Ningxin Dang, Jie Yu, Stephen J. Bush, Shenghan Gao, Wenxi He, Sirui Wang, Hongtao Guo, Xiaofei Yang, Weimin Ma, Kai Ye BMC Genomics . 25, 356 (2024). [PDF] We constructed near telomere-to-telomere (T2T) assemblies for two Trebouxiophyceae species, Chlorella sorokiniana NS4-2 and Chlorella pyrenoidosa DBH, with chromosome numbers of 12 and 13, and genome sizes of 58.11 Mb and 53.41 Mb, respectively. We identified and validated their centromere sequences using CENH3 ChIP-seq and found that, similar to humans and higher plants, the centromeric CENH3 signals of green algae display a pattern of hypomethylation. Interestingly, the centromeres of both species largely comprised transposable elements, although they differed significantly in their composition. Species within the Chlorella genus display a more diverse centromere composition, with major constituents including members of the LTR/Copia, LINE/L1, and LINE/RTEX families. This is in contrast to green algae including Chlamydomonas reinhardtii, Coccomyxa subellipsoidea, and Chromochloris zofingiensis, in which centromere composition instead has a pronounced single-element composition. Moreover, we observed significant differences in ...... |
.jpg") |
De novo and somatic structural variant discovery with SVision-pro Songbo Wang, Jiadong Lin, Peng Jia, Tun Xu, Xiujuan Li, Yuezhuangnan Liu, Dan Xu, Stephen J. Bush, Deyu Meng, Kai Ye Nature Biotechnology . 43, 181–185 (2025). [PDF] Long-read-based de novo and somatic structural variant (SV) discovery remains challenging, necessitating genomic comparison between samples. We developed SVision-pro, a neural-network-based instance segmentation framework that represents genome-to-genome-level sequencing differences visually and discovers SV comparatively between genomes without any prerequisite for inference models. SVision-pro outperforms state-of-the-art approaches, in particular, the resolving of complex SVs is improved, with low Mendelian error rates, high sensitivity of low-frequency SVs and reduced false-positive rates compared with SV merging approaches.Here we propose SVision-pro, comprising two key modules: a sequence-to-image representation module encoding genomic features from two samples in a single image, from which a neural-network recognition module comparatively recognizes SVs as well as their intergenome differences. SVision-pro integrates SV detection and genotyping between ...... |
 |
Haplotype-resolved assemblies and variant benchmark of a Chinese Quartet Peng Jia, Lianhua Dong, Xiaofei Yang, Bo Wang, Stephen J. Bush, Tingjie Wang, Jiadong Lin, Songbo Wang, ..., Han Xia, Yuanting Zheng, Leming Shi, Yi Lv, Jing Wang, Kai Ye Genome Biol . 24, 277 (2023). [PDF] The long reads from the monozygotic twin daughters are phased into paternal and maternal haplotypes using the parent–child genetic map and for each haplotype. We also use long reads to generate haplotype-resolved whole-genome assemblies with completeness and continuity exceeding that of GRCh38. Using this Quartet, we comprehensively catalogue the human variant landscape, generating a dataset of 3,962,453 SNVs, 886,648 indels (< 50 bp), 9726 large deletions (≥ 50 bp), 15,600 large insertions (≥ 50 bp), 40 inversions, 31 complex structural variants, and 68 de novo mutations which are shared between the monozygotic twin daughters. Variants underrepresented in previous benchmarks owing to their complexity—including those located at long repeat regions, complex structural variants, and de novo mutations—are systematically examined in this study. Recent state-of-the-art sequencing technologies enable the investigation of challenging regions in the human genome and expand the scope of variant benchmarking datasets. Herein, we sequence a Chinese Quartet ...... |
.jpg") |
A pangenome reference of 36 Chinese populations Yang Gao, Xiaofei Yang, Hao Chen, Xinjiang Tan, Zhaoqing Yang, Lian Deng, Baonan Wang, Shuang Kong, Songyang Li, Yuhang Cui,..., Chinese Pangenome Consortium (CPC), Yan Lu, Jiayou Chu, Kai Ye, Shuhua Xu Nature . 619, 112–121 (2023). [PDF] Human genomics is witnessing an ongoing paradigm shift from a single reference sequence to a pangenome form, but populations of Asian ancestry are underrepresented. Here we present data from the first phase of the Chinese Pangenome Consortium, including a collection of 116 high-quality and haplotype-phased de novo assemblies based on 58 core samples representing 36 minority Chinese ethnic groups. With an average 30.65× high-fidelity long-read sequence coverage, an average contiguity N50 of more than 35.63 megabases and an average total size of 3.01 gigabases, the CPC core assemblies add 189 million base pairs of euchromatic polymorphic sequences and 1,367 protein-coding gene duplications to GRCh38. We identified 15.9 million small variants and 78,072 structural variants, of which 5.9 million small variants and 34,223 structural variants were not reported in a recently released pangenome reference1. The Chinese Pangenome Consortium data demonstrate a remarkable increase in the discovery of novel and missing sequences when individuals are included from underrepresented minority ethnic groups...... |
 |
HiCAT: a tool for automatic annotation of centromere structure Shenghan Gao, Xiaofei Yang, Hongtao Guo, Xixi Zhao, Bo Wang, Kai Ye Genome Biology . 24, 58 (2023) . [PDF] Significant improvements in long-read sequencing technologies have unlocked complex genomic areas, such as centromeres, in the genome and introduced the centromere annotation problem. Currently, centromeres are annotated in a semi-manual way. Here, we propose HiCAT, a generalizable automatic centromere annotation tool, based on hierarchical tandem repeat mining to facilitate decoding of centromere architecture. We apply HiCAT to simulated datasets, human CHM13-T2T and gapless Arabidopsis thaliana genomes. Our results are generally consistent with previous inferences but also greatly improve annotation continuity and reveal additional fine structures, demonstrating HiCAT’s performance and general applicability. HiCAT takes a monomer template and a centromere DNA sequence as inputs. There are two steps in HiCAT: generation of a block list and similarity matrix and mining of HORs. |
 |
Cellular heterogeneity and transcriptomic profiles during intrahepatic cholangiocarcinoma initiation and progression Tingjie Wang, Chuanrui Xu, Zhijing Zhang, Hua Wu, Xiujuan Li, Yu Zhang, Nan Deng, Ningxin Dang, Guangbo Tang, Xiaofei Yang, Bingyin Shi, Zihang Li, Lei Li, Kai Ye Hepatology . 76:1302–1317 (2022) . [PDF] We performed single-cell RNA sequencing (scRNA-seq) using AKT/Notch intracellular domain–induced mouse ICC tissues at early, middle, and late stages. We analyzed the transcriptomic landscape, cellular classification and evolution, and intercellular communication during ICC initiation/progression. We confirmed the findings using quantitative real-time PCR, western blotting, immunohistochemistry or immunofluorescence, and gene knockout/knockdown analysis. We identified stress-responding and proliferating subpopulations in late-stage mouse ICC tissues and validated them using human scRNA-seq data sets. By integrating weighted correlation network analysis and protein–protein interaction through least absolute shrinkage and selection operator regression, we identified zinc finger, MIZ-type containing 1 (Zmiz1) and Y box protein 1 (Ybx1) as core transcription factors required by stress-responding and proliferating ICC cells, respectively. Knockout of either one led to the blockade of ICC initiation/progression. Using two other ICC mouse models (YAP/AKT, KRAS/p19) and human ICC scRNA-seq data sets, we confirmed the orchestrating roles of Zmiz1 and Ybx1 in ICC occurrence and development. In addition, hes family bHLH transcription factor 1, cofilin 1, and ...... |
.jpg") |
SVision: a deep learning approach to resolve complex structural variants Jiadong Lin, Songbo Wang, Peter A. Audano, Deyu Meng, Jacob I. Flores, Walter Kosters, Xiaofei Yang, Peng Jia, Tobias Marschall, Christine R. Beck, Kai Ye Nature Methods . 19, 1230–1233 (2022). [PDF] Complex structural variants (CSVs) encompass multiple breakpoints and are often missed or misinterpreted. We developed SVision, a deep-learning-based multi-object-recognition framework, to automatically detect and characterize CSVs from long-read sequencing data. SVision outperforms current callers at identifying the internal structure of complex events and has revealed 80 high-quality CSVs with 25 distinct structures from an individual genome. SVision directly detects CSVs without matching known structures, allowing sensitive detection of both common and previously uncharacterized complex rearrangements. We developed an automated CSV detection and characterization method: SVision. It introduces a sequence-to-image coding schema, adapting variant detection to a problem that is amenable to deep-learning frameworks. SVision contains three core components: an encoder that represents the differences and similarities between a variant-supporting read and its corresponding segment on the reference genome as a denoised image, a targeted multi-object recognition (tMOR) framework...... |
 |
Three chromosome-scale Papaver genomes reveal punctuated patchwork evolution of the morphinan and noscapine biosynthesis pathway Xiaofei Yang, Shenghan Gao, Li Guo, Bo Wang, Yanyan Jia, Jian Zhou, Yizhuo Che, Peng Jia, Jiadong Lin, Tun Xu, Jianyong Sun, Kai Ye Nature Communications . 12, 6030 (2021). [PDF] For millions of years, plants evolve plenty of structurally diverse secondary metabolites (SM) to support their sessile lifestyles through continuous biochemical pathway innovation. While new genes commonly drive the evolution of plant SM pathway, how a full biosynthetic pathway evolves remains poorly understood. The evolution of pathway involves recruiting new genes along the reaction cascade forwardly, backwardly, or in a patchwork manner. With three chromosome-scale Papaver genome assemblies, we here reveal whole-genome duplications (WGDs) apparently accelerate chromosomal rearrangements with a nonrandom distribution towards SM optimization. A burst of structural variants involving fusions, translocations and duplications within 7.7 million years have assembled nine genes into the benzylisoquinoline alkaloids gene cluster, following a punctuated patchwork model. Biosynthetic gene copies and their total expression matter to morphinan production. Our results demonstrate how new genes have been recruited from ...... |
 |
The opium poppy genome and morphinan production Li Guo, Thilo Winzer, Xiaofei Yang, Yi Li, Zemin Ning, Zhesi He, Roxana Teodor, Ying Lu, Tim A Bowser, Ian A Graham, Kai Ye Science . 362,343-347(2018) . [PDF] Morphinan-based painkillers are derived from opium poppy (Papaver somniferum L.). We report a draft of the opium poppy genome, with 2.72 gigabases assembled into 11 chromosomes with contig N50 and scaffold N50 of 1.77 and 204 megabases, respectively. Synteny analysis suggests a whole-genome duplication at ~7.8 million years ago and ancient segmental or whole-genome duplication(s) that occurred before the Papaveraceae-Ranunculaceae divergence 110 million years ago. Syntenic blocks representative of phthalideisoquinoline and morphinan components of a benzylisoquinoline alkaloid cluster of 15 genes provide insight into how this cluster evolved. Paralog analysis identified P450 and oxidoreductase genes that combined to form the STORR gene fusion essential for morphinan biosynthesis in opium poppy. Thus, gene duplication, rearrangement, and fusion events have led to evolution of specialized metabolic products in opium poppy. |


















